KẾT QUẢ GIỮA KỲ GIAI ĐOẠN 2 THỬ NGHIỆM LÂM SÀNG VẮC XIN COVIVAC DO IVAC SẢN XUẤT
Ngày 29 tháng 12 năm 2021, tại Bộ Y Tế, Hội đồng Đạo đức trong nghiên cứu y sinh học Quốc gia đã họp đánh giá kết quả giữa kỳ thử nghiệm lâm sàng giai đoạn 2 vắc xin COVIVAC do IVAC sản xuất. Giai đoạn 2 thử nghiệm trên 374 người tình nguyện tuổi từ 18-59 tuổi và 60 tuổi trở lên tại huyện Vũ Thư, tỉnh Thái Bình. Vắc xin COVIVAC cho thấy đạt yêu cầu về tính an toàn, được dung nạp tốt và sinh kháng thể trung hòa ở mức cao hơn so với vắc xin được cấp phép sử dụng khẩn cấp AstraZeneca. Những quan sát này làm cơ sở cho việc tiến hành thử nghiệm lâm sàng giai đoạn 3 để khẳng định tính an toàn và tính sinh miễn dịch vượt trội so với vắc xin được cấp phép sử dụng khẩn cấp Astrazeneca. Thông tin cụ thể được trình bày dưới đây.
Tên đề cương “Thử nghiệm lâm sàng giai đoạn 1 và 2, ngẫu nhiên, mù kép, có đối chứng với giả dược (giai đoạn 1) và với vắc xin COVID-19 đã được cấp phép (giai đoạn 2) để đánh giá tính an toàn và tính sinh miễn dịch của vắc xin COVIVAC do IVAC sản suất trên người trưởng thành từ 18 tuổi và từ 60 tuổi trở lên tại Việt Nam”
1. Vai trò chính của các tổ chức tham gia nghiên cứu giai đoạn 2
- Đề cương nghiên cứu được bộ Y tế phê duyệt ngày 09/08/2021
- Ngày tiêm mũi đầu tiên 18/08/2021, kết thúc tiêm mũi 2 ngày 20/9/2021
- Địa điểm nghiên cứu: Trung tâm Y tế huyện Vũ Thư, tỉnh Thái Bình
- Tổ chức nhận thử là Viện Vệ sinh dịch tễ trung ương (NIHE)
- Đơn vị phối hợp nghiên cứu giai đoạn 2: CDC tỉnh Thái Bình
- Chủ nhiệm đề tài (PI): GS.TS.Đặng Đức Anh, NIHE và PGS.TS.Vũ Đình Thiểm
- Đơn vị giám sát độc lập nghiên cứu trên thực địa: Vietstar, Việt Nam
- Đơn vị quản lý, phân tích dữ liệu độc lập: BIOPHICS, Đại học Mahidol, Thái Lan
- Phòng xét nghiệm miễn dịch: Nexelis, Laval, Quebec, Canada
- Phòng thí nghiệm vi sinh vật: Khoa vi rút, NIHE
- Tổ chức hỗ trợ kỹ thuật: PATH tại Việt Nam và tại Mỹ
- Nhà tài trợ: Viện vắc xin và sinh phẩm y tế (IVAC)
- Nhóm giám sát theo đề cương (PSRT), gồm 5 người (IVAC, PI, nghiên cứu viên, bác sĩ của Bệnh viện đa khoa tỉnh Thái Bình, PATH)
- Ban đánh giá an toàn và dữ liệu độc lập (DSMB): Gồm 4 chuyên gia
- Nhóm giám sát theo đề cương thực hiện việc giám sát định kỳ trong thời gian thử nghiệm
- Hội đồng đạo đức trong nghiên cứu y sinh học Quốc gia: Thực hiện việc kiểm tra, giám sát trong thời gian thử nghiệm.
2. Tóm tắt sản phẩm và thiết kế nghiên cứu giai đoạn 2
Vắc xin COVIVAC được IVAC bắt đầu nghiên cứu và phát triển từ tháng 5 năm 2020. Đây là dự án hợp tác với các trường Đại học của Hoa Kỳ - Trường Y Icahn tại Mount Sinai (Icahn Mount Sinai), nơi phát triển công nghệ vắc xin phòng COVID-19 dựa trên vi rút gây bệnh Niu-cát-xơn(NDV) tái tổ hợp và trường Đại học Texas ở Austin, nơi phát triển cấu trúc protein gai đột biến của vi rút SARS-CoV-2, HexaPro, nhằm tạo ra một cấu trúc bền vững và được sử dụng trong vắc xin- và tổ chức PATH. Từ chủng vi rút gốc giống được phát triển tại Icahn Mount Sinai, IVAC cùng với hai nhà sản xuất vắc xin khác ở Thái Lan và Bra-xin cùng nghiên cứu và sản xuất các liều vắc xin phòng COVID-19 NDV-HXP-S tái tổ hợp với sự hỗ trợ kỹ thuật của Icahn Mount Sinai và tổ chức PATH.
Các sản phẩm nghiên cứu trong giai đoạn 2:
- Vắc xin nghiên cứu COVIVAC: là vắc xin tái tổ hợp, toàn hạt vi rút, đã được bất hoạt. Vắc xin sử dụng vi rút gây bệnh Niu-cát-xơn (NDV), biểu hiện protein gai tái tổ hợp của SARS-CoV-2 (HexaPro S) trên bề mặt NDV. Liều dùng 0,5 ml chứa 3 µg protein S hoặc 0,5 ml chứa 6 µg protein S.
-
Vắc xin đối chứng AstraZeneca (AZD 1222): là một vắc xin véc-tơ vi rút, sử dụng adenovirus của tinh tinh đã mất khả năng sao chép, protein S của vi rút SARS-CoV-2 được các adenovirus chuyển đổi này đưa vào các tế bào của người tiêm vắc xin. Liều dùng 0,5 ml chứa 5 x 1010 hạt vi rút ChAdOx1-S.
Tóm tắt thiết kế nghiên cứu:
- Tổng số đối tượng tham gia nghiên cứu là 374. Các đối tượng nghiên cứu được chia thành 3 nhóm theo tỷ lệ 1:1:1, trong đó hai nhóm tiêm vắc xin COVIVAC với mức liều 3 µg hoặc 6 µg và 1 nhóm tiêm vắc xin đối chứng AstraZeneca (AZD1222) - Vắc xin được WHO cấp phép sử dụng khẩn cấp và đã được sử dụng tại Việt Nam. Các đối tượng được phân theo 2 nhóm tuổi từ 18-59 và ≥ 60 tuổi với tỷ lệ là 2:1.
- Phác đồ tiêm: Các đối tượng nghiên cứu được tiêm 2 liều vắc xin COVIVAC hoặc AZD1222 (0,5 ml/liều), cách nhau 28 ngày bằng đường tiêm bắp.
- Thu thập mẫu máu làm xét nghiệm miễn dịch được thực hiện trước tiêm mũi1, 14 ngày sau tiêm mũi 2 và 6 tháng sau tiêm mũi 2.
- Đo lường kháng thể trung hòa SARS-CoV-2 bằng xét nghiệm PNA50, chuẩn độ theo mẫu chuẩn của WHO (đơn vị tính IU/ml). Đáp ứng kháng thể IgG kháng protein S được đo lường bằng xét nghiệm ELISA, chuẩn độ theo mẫu chuẩn của WHO (đơn vị tính BAU/ml) và miễn dịch qua trung gian tế bào (Interferon gama và Interlekin 5) bằng xét nghiệm ELISPOT. Tất cả các xét nghiệm này được thực hiện tại Nexelis, Canada và kết quả miễn dịch được gửi trực tiếp đến tổ chức BIOPHICS ở Thái Lan để phân tích số liệu và viết báo cáo. Cả PI, IVAC hay PATH không được phép tiếp cận vào quá trình này.
- Đánh giá an toàn tại chỗ và toàn thân của các đối tượng sau tiêm 30 phút, trong vòng 7 ngày và 28 ngày sau mỗi mũi tiêm.
- Báo cáo kết quả giữa kỳ: Sau ngày D57 các số liêu an toàn trên thực địa được BIOPHICS rà soát, kiểm tra và đóng băng để cùng với kết quả miễn dịch từ Nexelis đưa vào phân tích và viết báo cáo giữa kỳ.
- Ban DSMB đánh giá độc lập dữ liệu về tính an toàn và gửi báo cáo trực tiếp cho Hội đồng đạo đức trong nghiên cứu y sinh học Quốc gia, gửi biên bản khuyến cáo về tính an toàn cho IVAC và PI.
3.Kết quả về an toàn
Tổng hợp kết quả về các biến cố bất lợi trong vòng 7 ngày và 28 ngày sau mỗi mũi tiêm
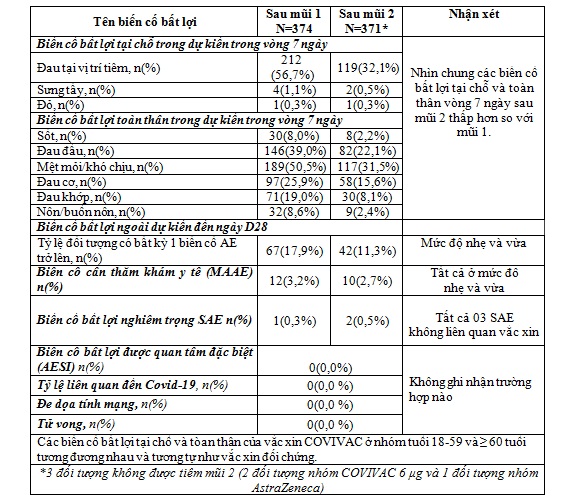
4.Kết quả về miễn dịch
Kháng thể trung hòa (NT50):
Mức độ kháng thể trung hòa được xem là có tương quan với hiệu lực bảo vệ của vắc xin. Nồng độ trung bình nhân (GMC) ở nhóm 3 µg và 6 µg của COVIVAC cao hơn nhóm vắc xin đối chứng và tương đương giữa nhóm tuổi 18-59 và nhóm tuổi ≥ 60 tuổi. Những quan sát này cần được khẳng định trong giai đoạn 3 với cỡ mẫu lớn hơn. Tỷ lệ đối tượng có GMC tăng ≥4 lần tại thời điểm 14 ngày sau tiêm mũi 2 so với trước tiêm đạt hơn 90 % ở nhóm vắc xin nghiên cứu (82,0% ở nhóm vắc xin đối chứng). Tỷ số tăng GMC ở thời điểm 14 ngày sau tiêm mũi 2 so với trước tiêm của nhóm vắc xin nghiên cứu đạt 33-39 (nhóm vắc xin đối chứng:17).
Miễn dịch qua trung gian tế bào: Tại thời điểm báo cáo giữa kỳ chưa có kết quả miễn dịch qua trung gian tế bào. Kết quả sẽ được cập nhật sau.
Dựa trên kết quả về tính an toàn và tính sinh miễn dịch của giai đoạn 1 và 2, vắc xin COVIVAC đủ điều kiện để chuyển sang nghiên cứu giai đoạn 3 với mức liều được khuyến cáo là 10 mcg.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}